Get Started¶
(All the code can be executed in the folder evoClustRNA/test_data/rp13 of this repository)
Prepare a multiple sequence alignment (MSA)¶
For the target sequence, the user needs to prepare an alignment or download it from the from the Rfam database. The sequence similarity should be reduced, using JalView to keep only diverse representatives. In theory, all sequences could be folded but because of the computational costs of simulations (6-10h per sequence for 80 CPUs, using either SimRNAweb or Rosetta FARFAR), we decided to fold only 4 the shortest sequences from the MSA. Once the final set of homologs to be folded was selected, the positions common to all sequences selected were determined.
 Figure 1. The alignment preparation. The conserved residues are marked with “x” in the pseudo-sequence “x”.
The marked as the conserved residues columns can be inspected in an arc diagrams of RNA secondary structures as the pink line (at the very bottom)
Figure 1. The alignment preparation. The conserved residues are marked with “x” in the pseudo-sequence “x”.
The marked as the conserved residues columns can be inspected in an arc diagrams of RNA secondary structures as the pink line (at the very bottom)
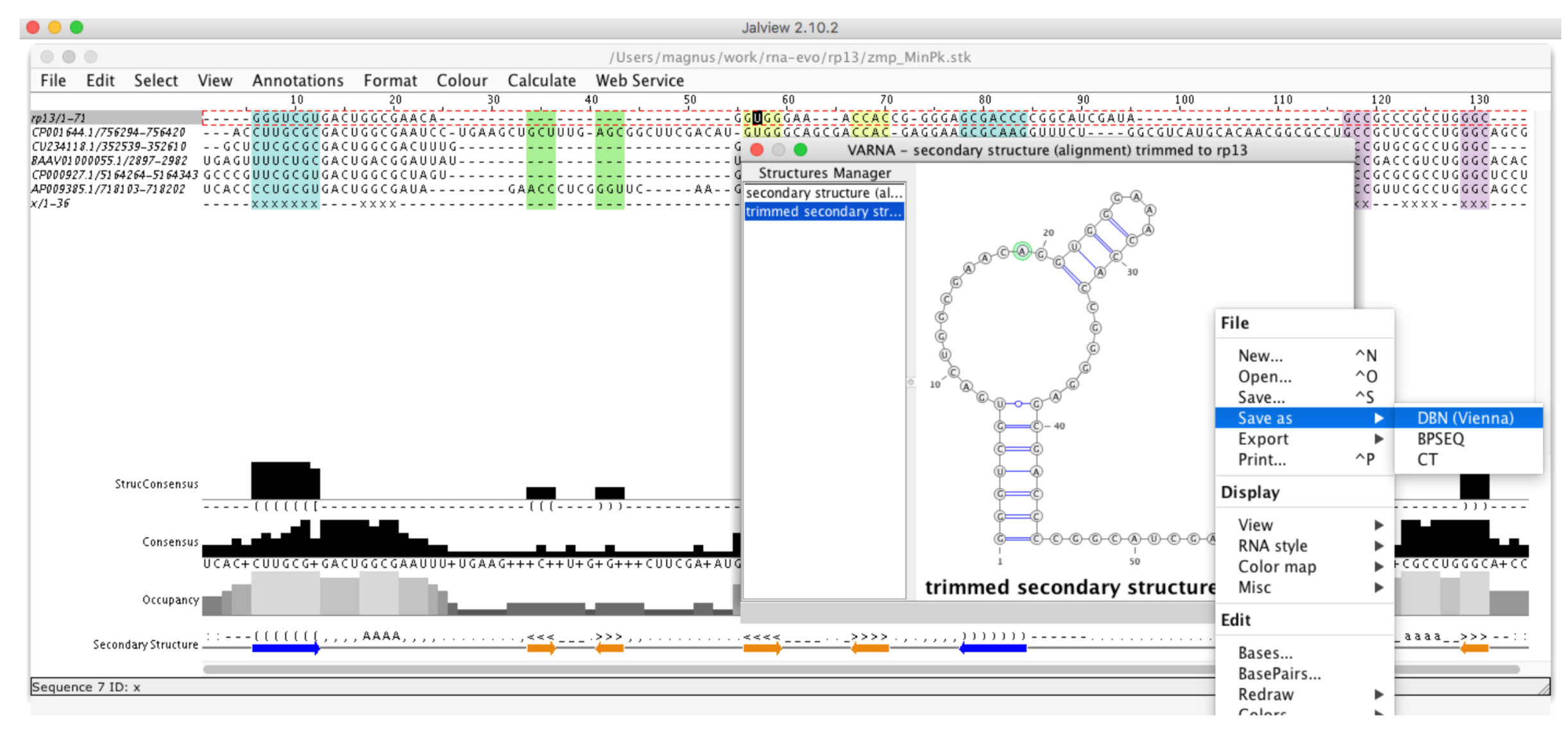 Figure 2. Generation sequence and secondary structure. Each sequence and associated secondary structure was “Saved as” to a Fasta file and used at the next stage of modeling with the use of the Jalview program.
Figure 2. Generation sequence and secondary structure. Each sequence and associated secondary structure was “Saved as” to a Fasta file and used at the next stage of modeling with the use of the Jalview program.
RNA 3D structure prediction to generate initial models¶
For each sequence chosen for folding, the user must prepare an input for RNA 3D structure prediction method. Two methods were used in this study: SimRNA and Rosetta. For Rosetta, a total of 10,000 decoys were generated for the target sequence and each homologous sequence using the Rosetta FARFAR protocol. For SimRNA prediction, SimRNAweb (https://genesilico.pl/SimRNAweb/) server was used using the default parameters.
To start with the EvoClustRNA protocol, we suggest using SimRNAweb first. The results are comparable to Rosetta, but the server is much easier to use for beginners.
At the end of modeling, top100 (or top200) models have to be extracted and copied into the structures folder.
Run EvoClustRNA clustering procedure (including extraction of conserved motifs)¶
Run evoClustRNA.py on an alignment (-a) and a folder with structures (-i) using mapping (-m) and flat directory structure:
[mm] evox$ git:(master) ✗ evoClustRNA.py -a ../rp13finalX_noSSperSeq_ref.sto -i structures -m ../mapping_ref.txt -f
\_ evoClustRNA Namespace(flat_dir=True, inf=False, input_dir='structures', mapping_fn='../mapping_ref.txt', matrix_fn='', output_dir='out', rna_alignment_fn='../rp13finalX_noSSperSeq_ref.sto', save=False, verbose=False)
rp13finalX_noSSperSeq_ref_mapping_refX.matrix
# of rnastruc: 6
rnastruc: ['rp13:tar_', 'rp13:solution', 'cp0016:zcp', 'nc9445:znc', 'nc3295:zc3', 'nzaaox:zza']
rp13 <-> tar_
cutting out fragments ...
analyzing... structures/*tar_*.pdb
# of structures 200
rp13 <-> solution
cutting out fragments ...
analyzing... structures/*solution*.pdb
# of structures 1
cp0016 <-> zcp
cutting out fragments ...
analyzing... structures/*zcp*.pdb
# of structures 200
nc9445 <-> znc
cutting out fragments ...
analyzing... structures/*znc*.pdb
# of structures 200
nc3295 <-> zc3
cutting out fragments ...
analyzing... structures/*zc3*.pdb
# of structures 200
nzaaox <-> zza
cutting out fragments ...
analyzing... structures/*zza*.pdb
# of structures 200
# of models: 1001
matrix was created! rp13finalX_noSSperSeq_ref_mapping_refX.matrix
evoClustRNA.py -a ../rp13finalX_noSSperSeq_ref.sto -i structures -m ../mapping_ref.txt -f
rp13finalX_noSSperSeq_ref_mapping_refX.matrix is the matrix with all-vs-all RMSDs for all conserved motifs.
Now it’s time to cluster the matrix:
evoClust_autoclustix.py rp13finalX_noSSperSeq_ref_mapping_refX.matrix
# of struc 1001
evoClust_clustix.py rp13finalX_noSSperSeq_ref_mapping_refX.matrix -c 0
n: 0
rm rp13finalX_noSSperSeq_ref_mapping_refX*cf0*.out # auto-removal
evoClust_clustix.py rp13finalX_noSSperSeq_ref_mapping_refX.matrix -c 0.5
n: 1
(....)
rm rp13finalX_noSSperSeq_ref_mapping_refX*cf8.5*.out # auto-removal
evoClust_clustix.py rp13finalX_noSSperSeq_ref_mapping_refX.matrix -c 9.0
n: 166
When the clustering is done, the best clusters can be obtained and copied to two folders for futher analysis: reps and reps_ns.
Copy the best cluster medoids from structures to reps
[mm] evox$ git:(master) ✗ evoClust_get_models.py -i structures/ *.out -u
evoClust_get_models.py
--------------------------------------------------------------------------------
1_tar_min.out.1.pdb
2_zcp_min.out.8.pdb
3_tar_min.out.66.pdb
4_tar_min.out.98.pdb
5_tar_min.out.25.pdb
= structures == out/structures/<files>===================
cp -v structures//tar_min.out.1.pdb reps/c1_tar_min.out.1.pdb
structures//tar_min.out.1.pdb -> reps/c1_tar_min.out.1.pdb
cp -v structures//zcp_min.out.8.pdb reps/c2_zcp_min.out.8.pdb
structures//zcp_min.out.8.pdb -> reps/c2_zcp_min.out.8.pdb
cp -v structures//tar_min.out.66.pdb reps/c3_tar_min.out.66.pdb
structures//tar_min.out.66.pdb -> reps/c3_tar_min.out.66.pdb
cp -v structures//tar_min.out.98.pdb reps/c4_tar_min.out.98.pdb
structures//tar_min.out.98.pdb -> reps/c4_tar_min.out.98.pdb
cp -v structures//tar_min.out.25.pdb reps/c5_tar_min.out.25.pdb
structures//tar_min.out.25.pdb -> reps/c5_tar_min.out.25.pdb
Copy the best cluster medoids from structures to reps_ns (this is where only models for the target sequences are stored, so no models of homologs):
[mm] evox$ git:(master) ✗ evoClust_get_models.py -i structures/ *.out -n tar -u
evoClust_get_models.py
--------------------------------------------------------------------------------
['tar_min.out.1.pdb', '', 'tar_min.out.66.pdb', 'tar_min.out.98.pdb', 'tar_min.out.25.pdb']
1_tar_min.out.1.pdb
2_
3_tar_min.out.66.pdb
4_tar_min.out.98.pdb
5_tar_min.out.25.pdb
= structures == out/structures/<files>===================
cp -v structures//tar_min.out.1.pdb reps_ns/c1_tar_min.out.1.pdb
structures//tar_min.out.1.pdb -> reps_ns/c1_tar_min.out.1.pdb
cp -v structures// reps_ns/c2_
cp: structures// is a directory (not copied).
cp -v structures//tar_min.out.66.pdb reps_ns/c3_tar_min.out.66.pdb
structures//tar_min.out.66.pdb -> reps_ns/c3_tar_min.out.66.pdb
cp -v structures//tar_min.out.98.pdb reps_ns/c4_tar_min.out.98.pdb
structures//tar_min.out.98.pdb -> reps_ns/c4_tar_min.out.98.pdb
cp -v structures//tar_min.out.25.pdb reps_ns/c5_tar_min.out.25.pdb
structures//tar_min.out.25.pdb -> reps_ns/c5_tar_min.out.25.pdb
Compare to the reference structure¶
OK, so now we have two folders with models that we can compare to the reference structure.
Various methods can be used to do that. For reps_ns (so the models for the reference sequence) you can use full atom RMSD:
[mm] evox$ git:(master) ✗ rna_calc_rmsd.py -t ..//*ref.pdb reps_ns/*.pdb
method: all-atom-built-in
# of models: 4
c1_tar_min.out.1.pdb 6.34 1295
c3_tar_min.out.66.pdb 11.6 1295
c4_tar_min.out.98.pdb 15.1 1295
c5_tar_min.out.25.pdb 14.34 1295
# of atoms used: 1295
csv was created! rmsds.csv
core RMSDs based on the alignment:
evoClust_calc_rmsd.py -a ..//*ref.sto -t ..//*ref.pdb -n rp13 -m ..//*mapping*ref.txt -o rmsd_motif.csv reps/*.pdb
Fri Jun 21 17:01:33 2019
Namespace(debug=False, dont_ignore_clusters=False, files=['reps/c1_tar_min.out.1.pdb', 'reps/c2_zcp_min.out.8.pdb', 'reps/c3_tar_min.out.66.pdb', 'reps/c4_tar_min.out.98.pdb', 'reps/c5_tar_min.out.25.pdb'], group_name='', mapping_fn='..//mapping_ref.txt', output_fn='rmsd_motif.csv', rna_alignment_fn='..//rp13finalX_noSSperSeq_ref.sto', target='..//target_13_solution_0_renumber_puzzle_ref.pdb', target_name='rp13')
target: ..//target_13_solution_0_renumber_puzzle_ref.pdb
# of rnastruc : 6
rnastruc: ['rp13:tar_', 'rp13:solution', 'cp0016:zcp', 'nc9445:znc', 'nc3295:zc3', 'nzaaox:zza']
WARNING: if any of your PDB file is missing, check mapping!
target model rmsd group_name
0 target_13_solution_0_renumber_puzzle_ref.pdb c1_tar_min.out.1.pdb 4.41
1 target_13_solution_0_renumber_puzzle_ref.pdb c2_zcp_min.out.8.pdb 16.08
2 target_13_solution_0_renumber_puzzle_ref.pdb c3_tar_min.out.66.pdb 10.32
3 target_13_solution_0_renumber_puzzle_ref.pdb c4_tar_min.out.98.pdb 15.50
4 target_13_solution_0_renumber_puzzle_ref.pdb c5_tar_min.out.25.pdb 15.24
or INFs:
[mm] evox$ git:(master) ✗ rna_calc_inf.py -f -t ..//*ref.pdb reps_ns/*.pdb
100% (4 of 4) |###########################################################################################################################################################################| Elapsed Time: 0:00:16 ETA: 00:00:00csv was created! inf.csv
[mm] evox$ git:(master) ✗ csv inf.csv
target fn inf_all inf_stack inf_WC inf_nWC sns_WC ppv_WC sns_nWC ppv_nWC
target_13_solution_0_renumber_puzzle_ref.pdb.outCR c4_tar_min.out.98.pdb.outCR 0.453 0.000 0.923 0.507 0.947 0.900 0.429 0.600
target_13_solution_0_renumber_puzzle_ref.pdb.outCR c3_tar_min.out.66.pdb.outCR 0.437 0.000 0.947 0.218 0.947 0.947 0.143 0.333
target_13_solution_0_renumber_puzzle_ref.pdb.outCR c1_tar_min.out.1.pdb.outCR 0.431 0.000 0.973 0.286 0.947 1.000 0.286 0.286
target_13_solution_0_renumber_puzzle_ref.pdb.outCR c5_tar_min.out.25.pdb.outCR 0.483 0.129 0.947 0.535 0.947 0.947 0.286 1.000
[Example of post-EvoClustRNA analysis]¶
The results can be also viewed with Clans, shown in the Figure 3 & 4.
In this clustering visualization, 100 models of five homologs are shown (each homolog uniquely colored, models of the target sequence are colored in lime). Models with a pairwise distance in terms of RMSDs lower than 6 Å are connected. The native structure was added to this clustering to see where it would be mapped. Interestingly, the native structure was mapped to the small cluster. In this cluster, there are three models for the target sequence. The model the closest to this the cluster center (Fig. 3B) achieved an RMSD of 6.98 Å to the native structure. This clustering visualization showed that there were models generated with the correct fold, but none of them were selected as the final prediction. The final prediction was the center of the biggest cluster (Fig. 3C).
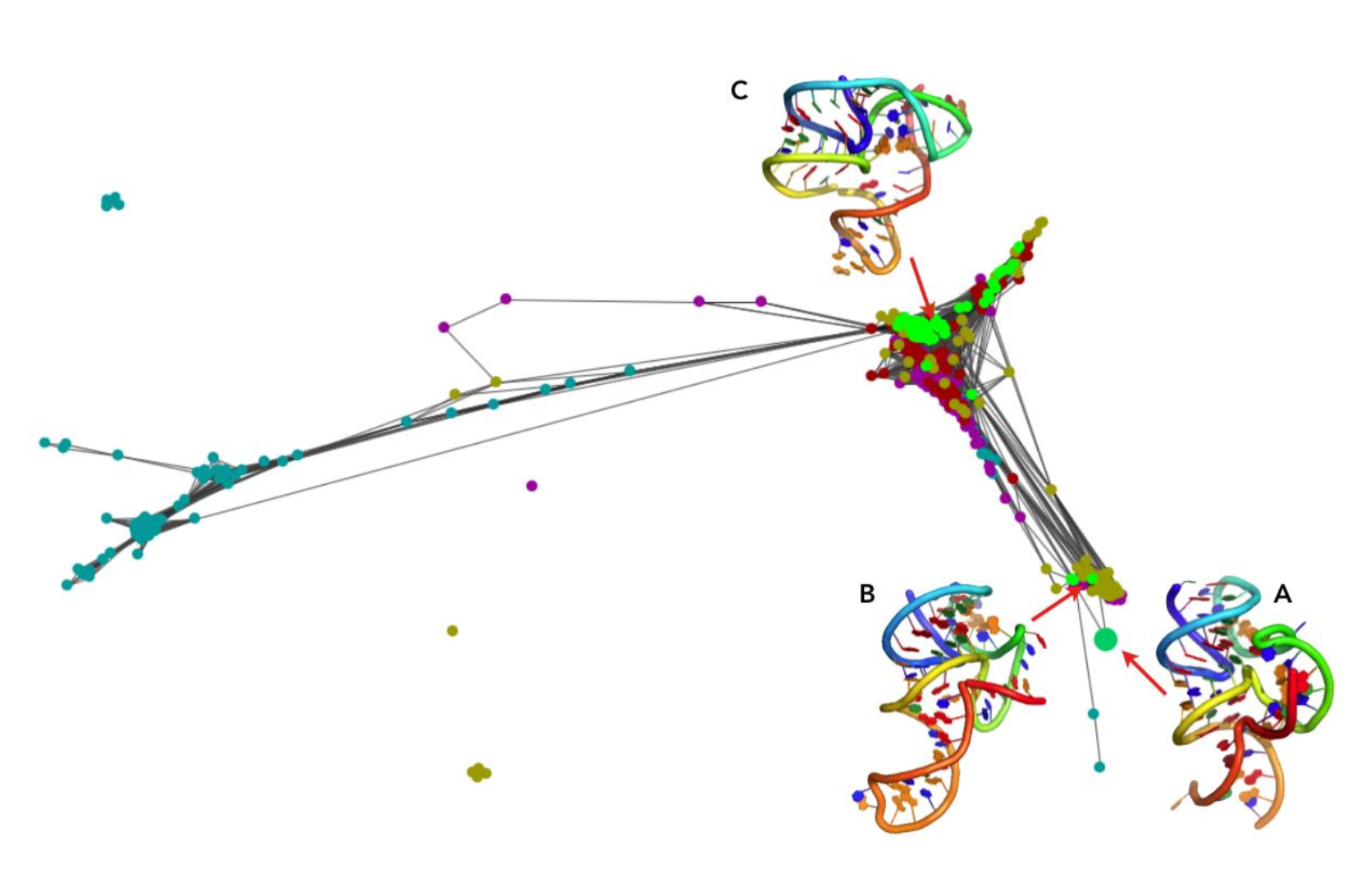 Figure 3. Clustering visualized with Clans for Pistol Ribozyme (RNA-Puzzle 17) (A) the native structure, (B) the model with the close fold to the native, detected in a small cluster, (C) the biggest cluster with the model that was returned as the final prediction.
Figure 3. Clustering visualized with Clans for Pistol Ribozyme (RNA-Puzzle 17) (A) the native structure, (B) the model with the close fold to the native, detected in a small cluster, (C) the biggest cluster with the model that was returned as the final prediction.
An analogous analysis was performed the results of clustering of EvoClustRNA|SimRNAweb run for the TPP riboswitch. Models with a pairwise distance in terms of RMSDs lower than 9 Å are connected. Interestingly, the native structure (Fig. 4A, big dot) was mapped to a cluster of models of one of the homologs (Fig. 4, blue). The center of this cluster (Fig. 4B) achieved an RMSD (of helical, shared fragments) of 9 Å to the native structure. In this cluster, there were not models for the target sequence. Since SimRNAweb was not able to detect non-canonical interactions, most of the structures were in “open” conformation and clustered far from the native structure. The final prediction was (Fig. 4C) achieved an RMSD of 24.08 Å with respect to the native.
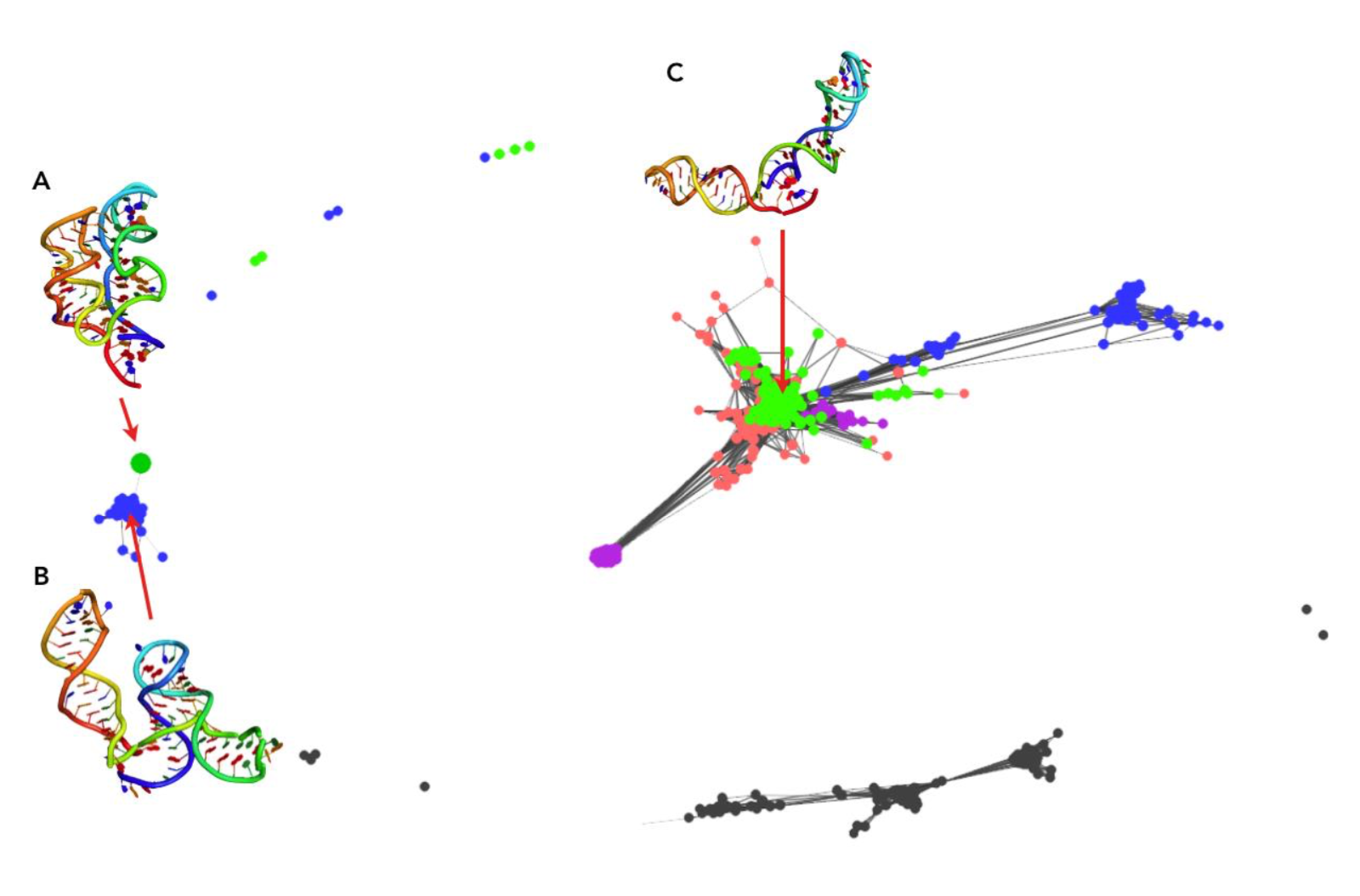
Figure 4: Clustering visualized with Clans for TPP riboswitch (A) the native structure, (B) the model with the close fold to the native (C) the biggest cluster with the model that was returned as the final prediction.
Learn more about Clanstix https://rna-tools.readthedocs.io/en/latest/tools.html#module-rna_tools.tools.clanstix.rna_clanstix
Figure 4: Clustering visualized with Clans for TPP riboswitch (A) the native structure, (B) the model with the close fold to the native (C) the biggest cluster with the model that was returned as the final prediction.